People

The Boss
Víctor de Lorenzo
I am a Chemist by training who was dragged into Molecular Microbiology and Environmental Biotechnology soon after being successively exposed to the beauty and power of the small in the Laboratories of Carlos Asensio (CSIC, Madrid), Tony Pugsley (Institut Pasteur, Paris), Joe Neilands (UC Berkeley) and Ken Timmis (GBF, Braunschweig). I have never repented of becoming a microbiologist of sorts and I hope that my longtime affair with Pseudomonas putida will last until the end! Follow me on Google Scholar.
You may download here my 1983 Thesis on Microcins
 |
 |
|---|---|
| Blurred 1982 picture with my Lab mates of the time I was making my PhD in Madrid (1982). The guy to the left was Joaquín Menéndez. Next is José García-Bustos, now Professor in Australia https://research.monash.edu/en/persons/jose-garcia-bustos. The lady was Amalia Montes, a wonderful technician now retired. Note Picasso’s Guernica posted on the wall, an icon of the Zeitgeist of the late 70s-early 80s of past century. | Victor working on his PhD Project (1983). Shaking water baths are now a rarity, but were very popular by that time. My supervisor (Carlos Asensio) died in an accident in 1982 and I had to finish my Thesis in a rush, basically on my own. I use to joke my students that I am the evidence that one does not need a supervisor to do a good PhD! My own experience also makes me a bit critical of the over-mentoring that newcomers expect these days. |

Executive Assistant
Inés Merino
I am the executive assistant of the Laboratory. Among my duties are the follow up of the Lab projects, organization of workshops and scientific meetings.

Technician
Sofía Fraile
I am the lab technician. I have a degree in Biological Sciences from the Autonoma University of Madrid and I have been in the laboratory for over 25 years tuning up multiple techniques of molecular biology. Among other tasks, I keep the stock of laboratory strains plus the new pSEVA collection. “I am the person who knows where all things in the lab”.
In the last time, we are looking for different strategies to create artificial communities of bacteria modifying the adhesive properties of P. putida. This could have an enormous biotechnological potencial.
I created in the lab the nanoPad platform, an important factory in order to produce and select highly specific adhesins. These adhesins are monomeric recombinant antibodies or nanobodies. The nanobodies against different proteins are using as tools to detect and manipulate different biological activities as well as the adhesive properties of P.putida.
P.putida, our bacteria model due to its plasticity; its capacity to use different organic compounds as carbon sources, including important contaminants such as lindane, toluen, etc.
These monomeric recombinant antibodies can be packaged into phage M13 to achieve new rounds of panning, secreted into the extracellular medium, expressed intracellularlly or bacteria surface expression, allowing us the binding with other bacteria or solid surfaces.
In the laboratory, we also have external membrane anchoring systems that can express surface proteins such as intimin and autotransporters.
The last technique that I used to generate specific adhesion is QCM, quartz crystal microbalance. This allows us to see if there is physical binding between bacteria and substrate with a serie of advantages: I deposit the substrate in the crystal with the gold in a vertical position and create a constant flow of bacteria to prevent the non-specific binding of these.
QCM measures mass-frequency relationship. Frequency change is translated in an increase in the thickness.
In the future, we are planning to select VHHs recognizing different proteins from strains of interest, in order to generate Bacterial Consortia.
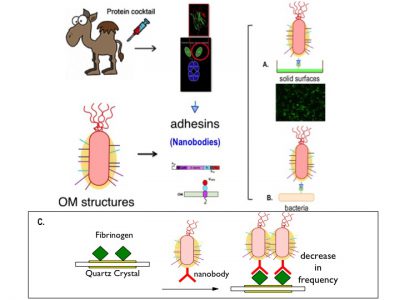
Schematic representation of the use of nanobodies as adhesins in P. putida. The OM anchoring system used is intimin. A. Bacteria binding to a substrate or solid surface. B. Adhesión bacteria with other bacteria.
- Detection of nanobodies again fibrinogen. Only those bacteria that express this nanobody will stick, thus increasing the mass deposited on the crystal and lowering the frequency.
See more in: Fraile S et al. Proteomics. 2013 Oct;13(18-19):2766-75.

Associated Researcher
Belén Calles
My research interests are mostly related to regulation of biological processes. In particular, my Ph.D. research was about different mechanisms of prokaryotic regulation of transcription, using Bacillus subtilis phage phi29 as model system. More recently, I have contributed to the study of the regulation of gene expression by the cAMP-CRP system and the role of the major RNA chaperone Hfq in the soil bacteria Pseudomonas putida.
Understanding biological processes is also important for Synthetic Biology applications to manipulate bacteria to perform novel complex behaviors. New organisms that satisfy human needs can be constructed by re-progamming extant cellular pathways to produce added value compounds. A key problem in such approaches is isolating specific metabolic pathways away from their side reactions.
To address these questions, we have developed a Tn5 based-transposon designed for the functionalization of target proteins with new traits. The tool enters in-frame peptides of different sizes within permissive regions with high efficiency. The system allows saturating the gene of interest with insertions that can be then screened to select for the desired knock-in into the protein of interest, without loss of the original functionality. One particular transposon variant was designed to produce GFP sandwich fusions and/or to insert very specific NIa protease cleavage sites, producing conditional knock-outs. Induction of the cognate protease cleaves and switches-off NIa tagged enzyme(s), causing a proteome rearrangement which in turn re-directs metabolic flux in a rational way. In addition, we have explored the possibility of producing transcriptional factors that not only respond to its natural inducer but also to the protease, which changes its logic operation by cleaving different parts of the regulatory protein. This transposon-tool can also be used for the functionalization of proteins with any other new trait of interest.
Another important issue when engineering new genetic circuitry is the need of optimized and well characterized regulatory nodes that control gene expression. Regulation of gene expression at appropriate times is, for example, crucial to avoid metabolic burden and toxicity when using cell factories as production platforms. By taking advantage of our standardized SEVA platform we have made a systematic study and comparison of the performance of the expression systems available to date, that will be very useful when selecting the most appropriate regulator/promoter pair.
To further improve fine-tuning of gene expression we have developed a translational regulatory tool that can be combined with different transcriptional regulation systems allowing tight control in Gram-negative bacteria, e.g. E. coli and Pseudomonas putida. The engineered module includes a sRNA that inhibits translation of leaky mRNAs. Expression of this regulatory sRNA is controlled by a cross-inhibition bistable circuit. Genes controlled under this regulatory circuit showed negligible leaky expression. This feature was instrumental to clone highly toxic colE3 colicin gene in the absence of the cognate immunity protein. In addition, the system showed a clear reduction of cell-to-cell variability. A further benefit of this gene expression device is that it could be integrated under the control of virtually any positive or negative transcriptional regulation system as a plug and play circuit.
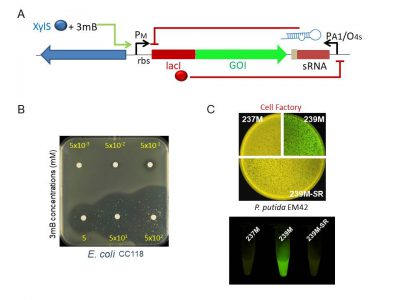
Schematic representation of the regulatory device tailored to have an all-or-none expression behavior of the gene of interest. The engineered module, which is SEVA-platform compatible, includes a sRNA that inhibits translation of leaky mRNAs. Expression of this regulatory sRNA is controlled by a cross-inhibition bistable circuit (A) Top agar plates with Escherichia coli cells transformed with the regulatory device harboring the highly toxic colE3 colicin, without the cognate immunity gene under the control of the XylS/Pm transcriptional regulation system. Note that induction of the expression of the colE3 gene with 3-methilbenzoate (3mB) kills the bacterium even at low concentrations (50 mM) (B). Basal expression of GFP cloned in AlkS/pAlkB regular expression system (239M) or the same transcriptional control in combination with the translational regulatory device (239M-SR) in Pseudomonas putida EM42 strain (the so called cell factory) in solid agar plates and liquid cultures. Note that 237M cells, which harbor the plasmid backbone with GFP but no expression system, were used as control of autofluorescence.

Postdoctoral Researcher
Esteban Martínez García
I obtained my PhD in Biology at Universidad Complutense in Madrid (UCM) in 2001, where I studied the bacterial physiology of stationary phase under the supervision of Antonio Tormo and Juana María Navarro. Then, I stayed until the end of 2002 as a postdoc in the laboratory of Julián Perera (UCM) working on bacterial biodegradation. Next, I moved to the U.S.A for a postdoctoral position. First, I worked on comparative genomics of Pseudomonas aeruginosa for three years in Roberto Kolter’s laboratory at the Harvard Medical School. Then, I moved to Kevin Foster’s lab at Harvard University where I worked on social interactions of P. aeruginosa.
After that period, I returned to Spain and in 2008 started working in Víctor de Lorenzo’s laboratory at CNB where I focused on different aspects related to environmental microbiology and synthetic biology. Doing that, we realized about the lack of efficient genetic tools to properly work with environmental bacteria. Along that line, part of my work is to develop a collection of modular tools for deployment of complex phenotypes in bacteria (SEVA collection). Also, I am especially motivated to adopt emerging techniques, such as recombineering and CRISPR/Cas9, to powerfully engineer bacterial genomes. All of these efforts are intended not only to broad the toolbox repertoire but also to facilitate adopting standards in synthetic biology that would help to efficiently program biological systems with new properties.
In the lab, we focus our work to Pseudomonas putida because, we believe, is an organism with a great potential for biotechnology and synthetic biology. The two most important facts that contribute to that potential are its huge metabolic versatility and not being a pathogen.
Particularly, we would like to blend the new developed tools with the knowledge gained through basic science to construct different bacterial chassis that enhance the natural performance of P. putida in different biotechnological and environmental tasks.
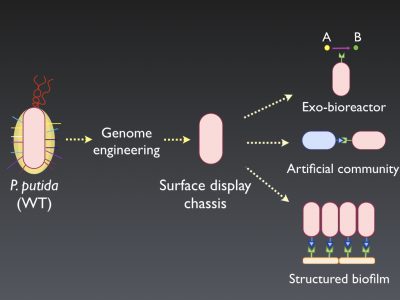
In order to do that, we are mainly developing two types of bacterial chassis: (i) cell factories and (ii) surface display strains. The first type comprises developing bacterial strains that have been manipulated to centralize its energy and reducing power into the production of interesting added-value metabolites and to be laboratory-friendly, meaning that are easy to perform common genetic manipulations. The second type, involves the modification of the natural adhesive properties of P. putida by developing a “naked” variant to efficiently display different adhesins to re-direct its attachment in a controlled fashion, being able, in this way, to engineer artificial communities, exo-bioreactors, or to build structured biofilms on surfaces (Figure 1).
Legend to figure
Schematic representation of the different utilities of the surface display chassis strain.

Postdoctoral Researcher
Yamal Al-ramahi
I received my PhD in Microbiology from the Autonomous University of Madrid. My PhD project was focused on following a Protein Engineering approach to expand the fluorescent protein “color palette” suitable as cellular localization tools in thermophiles. First, rational design was used to obtain cyan, cyan-green, green and yellow variants from the scaffold of superfolder GFP. Second, a method of directed molecular evolution by random mutagenesis allowed to obtain libraries to screen for derivatives of mCherry. This work yielded several proteins that were characterized and validated as localization tools in Thermus thermophilus. In the current episode of my scientific adventure at Víctor de Lorenzo´s laboratory in the National Center of Biotechnology, I am developing a platform based on Synthetic Biology to obtain variants of Antibodies.
Since my incorporation to Victor de Lorenzo´s laboratory I am nourished with a broad repertoire of resources that I can use to develop new molecular technologies hosted in microbes. In this context, my main project at Victor de Lorenzo´s laboratory aims at creating a device able to work in vivo to generate diversification of specific target sequences of interest while the accumulation of undesired mutations in the host chromosome is prevented.
Antibodies are proteins produced by the immune system and are very efficient at binding molecules specifically. Although this property makes them the tools of choice in a wide number of applications, both antibodies and antigen-binding fragments are difficult and expensive to produce with current procedures. This is the reason why we set out to design and implement an alternative technology to produce antibodies in a fashion reminiscent of the immune system but assembled with Synthetic Biology approaches. To this end, we embarked in setting up a platform made of parts assembled in bacteria that behaves as an immune-system-like machine. With this setup, single-chain camel antibodies (nanobodies) are generated and displayed on the bacterial surface—thus enabling a large number of uses in medical and industrial Biotechnology. First, autonomous functional modules are assembled in the genomic chassis of an Escherichia coli strain to allow the display of nanobody scaffolds on the surface of the cell. Second, the conditional diversification with mutagenesis of target-binding sequences of the nanobody lead to affinity maturation. Finally, binders with improved affinity and specificity towards desired molecular targets are selected. The hereby described artificial immune system (ARISYS) will dramatically simplify the production of recombinant antibodies.
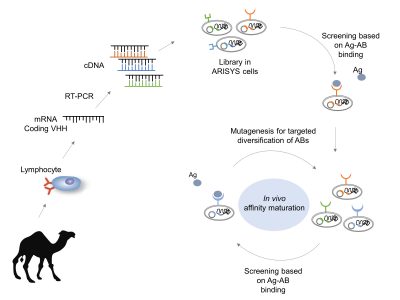
ARISYS: Artificial Immune System. Nanobodies from camels are light antibodies consisting on one heavy variable region (VHH). Libraries of vhh genes are cloned in bacteria. Only the ARISYS clones that specifically bind the antigen of interest are selected. Then, the genetic diversity-generating device mutates this vhh in order to generate a new library ready for a new maturation step. In the end, there are clones with greater specific binding ability against the antigen of interest

PhD Student
Angeles Hueso-Gil
My name is Angeles Hueso-Gil and I am doing my PhD at Victor’s lab. I was always interested in life sciences, so studied Biology and a Biotechnology master in Valencia, my hometown, and later I moved to Madrid to make my PhD.
My PhD project consists of the development of several tools and devices that will be placed into the ARYSIS circuit, which has the aim of producing affibodies (the variable part of camel antibodies) inside bacteria as Pseudomonas putida. In this circuit, the affibodies will be produced and diversified in a similar way that happens inside mammal cells, getting as a result a final affibody with a higher affinity for the antigen.
I have three main tasks involved with this project.
1-Developing a reversible light switch to control the conditional diversification of the affibodies. For this purpose I used the CcaS-CcaR green light system from Synecocystis, which was previously adapted to Escherichia coli. After several mutagenesis cycles of the construction carrying all the CcaS-CcaR components, we selected an optimized clone for P. putida and we are using it for its implementation into the ARYSIS circuit and the control of biofilm production.
2-Developing a surface contact switch which triggers the mechanism of the circuit once bacteria find their antigen and attach to the surface. I am characterizing the transcriptomic response of P. putida to solid surfaces and some of its natural contact systems of P. putida, as for example wsp complex, in order to know about possible tools suitable for our needs.
3-Expression of proteoRhodopsin and the production of its cofactor, the retinal, inside P. putida. This transmembrane protein is able to transform light energy into a proton gradient that will be used for ATP production inside the bacteria. The idea is to increase the energy supply inside our bacteria for the new functions of the ARYSIS circuit using the proteoRhodopsin activity.
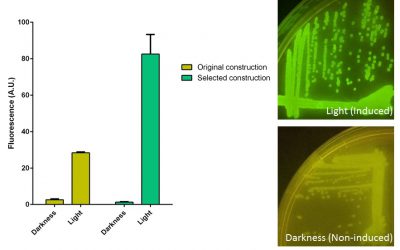
Behaviour inside Pseudomonas putida of the original construction carrying the CcaS-CcaR system and the selected clone after diversification. GFP production is induced when cultures

Postdoctoral Researcher
David Rodríguez Espeso
My name is David. I love science, but are the real science applications of it what really fascinate me. For that reasion my basic background is Chemical Engineering, but I did my PhD in applied mathematics (modelling and computation of bacterial biofilms).
I decided to work in the biology field with the initial purpose of finding a way to bring together biology and engineering by developing integrative solutions which take profit from both fields.
The use of microorganisms at industrial scales involves to know how bacteria work at a level of community: spreading, differentiation, morphogenesis or interaction with the physical boundaries are different aspects where there is a large gap of knowledge in many aspects that limit their application. This is my natural niche of work: understand the interactions between bacterial communities and the different physical and chemical phenomena developed inside and outside of those, with the purpose of gaining control of certain properties that allow us to use them in real applications.
Currently I’m focused on three main issues related with the organization and handling of bacterial systems: how bacterial colonies organize naturally in different physical media, how to modify bacterial biofilms artificially and how can we modify the organization of bacteria at individual level.
In the first issue we aim at acquiring a fundamental intuition about what kind of physical and chemical processes are taking place inside a bacterial biofilm when bacteria are left to evolve naturally in different systems (i.e channels, surfaces, flow, etc.).
Engineering bacterial biofilms for using them as real bioreactors is another of my projects: I’m attemping to gain control about the geometry and properties of a biofilm in order to modulate certain desired chemical reactions performed by bacteria, thus turning biofilms into micrometric controllable self-sustainable reactors.
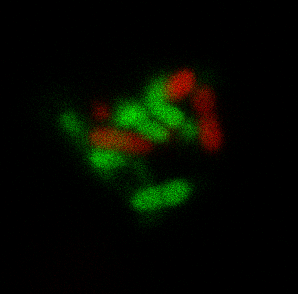
Finally my main third project is focused on studying how to create complex bacterial macrostructures starting from individual bacteria. By analysing growth processes of bacterial packs and assuming certain hypothesis about ordered movement of bacteria or fixing certain assembly rules, we aim at gaining insight about what could be the final obtained structures and thus helping to create self-assembly pathways that allow the creation of bacterial clusters with artificial guided patterns specifically designed for biotechnological applications (i.e. catalytic platforms).
 |
 |
Figures: a) CLSM image of a self-assembling bacterial aggregate designed for enhancing biochemical transformations of interest. b) Automated fluidic platform connected to a 3D printed turbidostat to perform evolution experiments.

Technician
Tomás Aparicio Pérez
I got a PhD in Biology under the supervisión of Dr. Perera (Faculty of Biology-UCM, 2000), where I studied the biology of a Thiobacillus plasmid. After that my profesional track included experience in a biotech company (PharmaMar S.A.-Madrid, 2001-2004), technical work in the Unit of Genomics of Scientific Park of Madrid (PCM, 2004-2009), a post-doc period in the CBMSO (Madrid, 2010) under the supervisión of Dr. García-Ballesta and also teaching activities as Associate Professor in UCM and UFV universites. Although involved in different research fields along my career, I have been mainly focused in Microbiology and Bacterial Genetic Engenieering.
In Victor´s Lab (CNB-Madrid, 2013-), I have a technical position aimed to give support for general laboratory activities, focusing on P. putida KT2440 strain engineering and pSEVA platform development. In this regard I colaborate in a number of specific projects such as recombieering and CRISPR/Cas9 assisted GE, Yeast Assembly, contruction of a variety of shuttle vectors (Gram+, yeast, streptomyces) and new expression systems design.

PhD Student
Elena Velázquez
My name is Elena Velázquez and I studied Biotechnology at the Technical University of Madrid (UPM). The reasons for this were my desire to understand how living organisms work and be able to tune them for something useful and new. That is why, after hearing the term “Synthetic Biology”, I was really attracted to this field. At this point, I was lucky enough to be accepted in Victor’s lab to perform my master thesis and, finally, I decided to start my PhD with a FPU fellowship.
In the lab, I have two main goals:
- Development of a biosensor able to detect 2,4-Dinitrotoluene (2,4-DNT): This compound is one of the main degradation products of anti-personnel landmines. In order to do this, I am currently implementing in Pseudomonas putida a technology based on a cytosine deaminase fused to the T7 RNA polymerase. This deaminase mutates DNA by turning cytosine into uracil, which is finally fixed as adenine in the DNA strand.I plan to use this fusion protein as the driving force to alter the specificity of a transcriptional factor in P. putida and find a version which is able to detect 2,4-DNT at high efficiency.
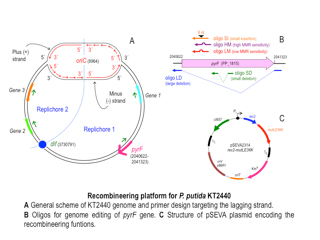
- Setting of a DNA-barcoding platform: In collaboration with the group of Dr. Natalio Krasnogor in Newcastle, we are developing a system meant to insert a particular, short and traceable sequence into the bacterial genome. This fragment of DNA codifies important information that you could seek for in a data base. With this purpose, we have engineered a group II intron from Lactococcus lactis to bear this “barcodes” and deliver them to precise sites of the chromosome.
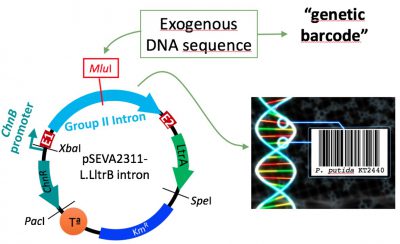
Figura 1. Schematic representation of our barcoding platform. From left to right: expression plasmid bearing the group II intron with the “DNA-barcode” inserted at MluI site. After induction, this group II intron integrates into the selected locus, labelling the strain.
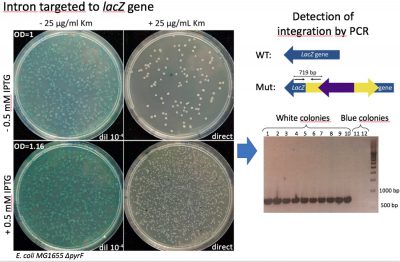
Figure 1. Group II intron integration assay. After induction of group II intron expression, this molecule is able to recognize lacZ gene sequence and insert in the designated place. After integration, the kanamycin resistant gene inside of it is activated and insertion mutants can be selected by plating the culture in the presence of this antibiotic. (A) Left plates: viability check. Right plates: selection of integration mutants. White colonies, a priori, have the group II intron inserted in the right position, disrupting lacZ gene. Blue colonies resistant to kanamycin are off-target integration mutants, with the intron inserted in other parts of the genome. (B) Test of correct integration through PCR: The presence of the intron in the correct position of lacZ gene produces a 719 bp amplicon while its absence leads to not amplification.

Technician
Elena Algar
I am a biologist who has always been enthusiastic about applied research. I obtained my PhD (2011) within the “Biotechnology of the rhizosphere” group lead by F. Javier Gutiérrez Mañero (Universidad CEU San Pablo, Madrid). In that period, I participated in several R&D projects related to different ways to improve plant-microbe interactions through various biotechnological applications, such as the exploitation of metagenomics to discover new genes and enzymes, to increase the production of interesting bioactive compounds, biofertilizers, biocontrol agents. During that time I spent four months in the Austrian Centre of Industrial Biotechnology (ACIB GmbH, Graz, Austria).
After my PhD I completed a Master in Biotechnology, (2016, Aliter-Escuela Internacional de Negocios de Madrid) oriented to gain expertise in the management part of science.
Then, I joined Victor’s group to contribute to the molecular tool repertoire and to exploit and engineer P. putida KT2440 to produce new chassis with increased performance for synthetic biology.
More specifically, we have developed new SEVA plasmids that will expand the already established collection. Moreover, we followed the SEVA standard to construct a new broad-host-range cosmid which can be used to carry out functional screenings of metagenomics libraries in different hosts.
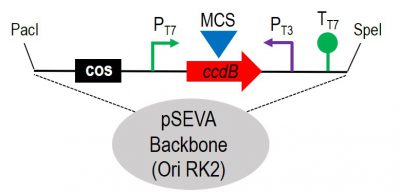
Figure 1. Schematic representation of the cargo region of the broad-host-range SEVA cosmid vector with the RK2 replicon. The COS site allows phage packaging of the DNA, while the multicloning site (MCS) is placed into the “Killer” gene ccdB allowing us to minimize the selection of empty vectors. Functional screenings could be done by inducing the expression of the PT7/ PT3 promoters and its transcriptional termination is enforced by the presence of the Tt7 and the T0 of the SEVA backbone, respectively. The system has the SEVA format with the advantages of its modularity.
On the other hand, I also have two basic science related projects associated to P. putida KT2440. However, we plan to use that knowledge to minimize horizontal gene transfer (HGT) and to increasing resistance to desiccation of this bacterium.
For the first purpose, we constructed a double ΔungΔdut mutant strain, which has its DNA enriched in uracil. We expect that when a strain with a high uracil content transfers plasmid DNA (U enriched) to a wild type host, that DNA would be degraded by the host base excision repair mechanism. Ideally, that double mutant strain could be used as a “biosafe strain” to minimize horizontal transfer events.
The second topic is related to desiccation stress in KT2440. First, by studying the desiccation stress response of this bacterium, and particularly by the characterization of a promoter (P4707) which specifically responds to desiccation conditions. Finally, we would like to endow P. putida with a higher resistance to desiccation by heterologous expression of genes coming from different organisms (tardigrades, Deinococcus, Arthrobacter…) that naturally have a great potential to endure those conditions.

Figure 2. Representative image of the P4707 characterization in P. putida KT2440. The image shows a colony section (xy plane) of KT2440 taken by a confocal microscopy. KT2440 constitutively expresses cherry and harbors a plasmid with a transcriptional fusion of the P4707 promoter to GFP (pSEVA237 plasmid), growing on agarose plates with low humidity (less than 30% RH) for 48h. From left to right, GFP channel, mCherry channel and merged image, where the xz plane is also shown at the bottom. The specific response of that promoter (GFP) to dessication conditions is shown in the left picture of the image.

PhD Student
Huseyin Tas
I received my B.Sc. in Biological Sciences and Bioengineering at Sabanci University, Turkey (2012) and received my M.Sc. in Biophysics and Computational Biology at University of Illinois Urbana-Champaign, USA (2014). During my Masters, I worked on developing genomic manipulation techniques for precise bacterial genome editing (for details see also: Tas et al, PLoS One, 2015). Having completed my Masters, I have become more interested in Synthetic Biology and its applications and now I am doing my PhD studies in this field under the advisory of Prof. Víctor De Lorenzo at Consejo Superior de Investigaciones y Ciencias (CSIC, Madrid).
In De Lorenzo Lab, I am mainly working on programming bacterial genome (specifically Pseudomonas putida) in order to apply this concept for environmental and industrial processes. This concept includes building artificial pathways and organizational circuitries in the bacterial genetic information in order to provide Output-of-Interest (can be bioremediation, production of a industrially important side or raw materials etc). This is done through implementing genetic circuits based on Boolean logic. The concept is not only important for incorporating the idea of biosensors in an applicable platform especially for the environmental systems, but also important for the proof of concept for the future programmable robotic bugs via showing interoperability of biosensors and/or building blocks of the circuits.
However, this system has some bottlenecks. Synthetic Biology is a field that has been an attention magnet especially for the last two decades. Although, there have been numerous valuable inputs into the field, still we are lacking of having better standards in order to build comprehensive systems. This is in mind, I am working on Standardization of synthetic biology. As the motivation of having standards is to enable synthetic biology research to be a robust platform for genomic modifications, my aim is to bring universal standards for synthetic biology via using Pputida as the model organism.
In addition to these, being aware of the fact that pharmaceutical industry is encountering several bottlenecks one of which is having high purity raw materials and host-immune reactive down-processing agents, I am developing a microbial cell factory that is specifically designed for pharmaceutical industry usage to lessen and overcome these bottlenecks in the filed.
To sum up, having a standardized platform furnished with point-of-interest biosensors integrate-able to Pputida that can sense, process and react to its environment is my ultimate goal.
For further details about myself, you can also check my personal web page.
